
{kind=link}
На рисунке изображена гипотетическая схема действия фермента SpnF. В сужающийся канал с одного конца (на рисунке справа) попадает молекула реагента (субстрата), а с другого конца (слева) выходит молекула продукта, отличающаяся от исходной молекулы реагента наличием двух дополнительных связей (нарисованы белым), замыкающих цикл. Изображение © Лаборатория В. Ананикова
Долгое время считалось, что реакция циклоприсоединения (реакция Дильса–Альдера) является исключительно синтетическим методом органической химии и не встречается в природе, поскольку не было достоверных доказательств существования ферментов, катализирующих такой процесс. Интереснейшей находкой оказалась бактерия Saccharopolyspora spinosa: этот микроорганизм, как выяснилось, способен осуществить внутримолекулярную реакцию циклоприсоединения, позволяющую получать полициклические биологически активные продукты. В результате теоретического исследования, выполненного в Институте органической химии имени Н. Д. Зелинского РАН (Москва), предложена модель, позволяющая объяснить механизм этого уникального процесса. Результаты исследования, опубликованного в журнале PLoS ONE, дают новое представление о возможном механизме действия ферментов в таких процессах.
Катализаторы: преодолевая барьеры[]
Любой живой организм в своей материальной основе — это сложноорганизованная химическая система, устойчивое существование которой возможно благодаря огромному множеству согласованных друг с другом за миллионы лет эволюции биохимических процессов, протекающих в его клетках или клетке. Чтобы выжить, организм должен реагировать на быстро изменяющиеся условия окружающей среды. Для этого требуется, чтобы биохимические реакции протекали с высокой скоростью.
Как известно, многие химические процессы могут быть ускорены во много раз с помощью специальных веществ — катализаторов, а само явление такого ускорения называется катализом. Катализатор — это вещество, которое направляет химический процесс превращения субстрата (то есть исходного реагента) в продукт по пути, требующему для свой реализации меньше энергии. В результате этого и происходит ускорение реакции по сравнению с некаталитическим процессом. Активно участвуя в химической реакции, катализатор восстанавливает свою структуру после ее завершения и готов снова выполнять свою ускоряющую работу. То есть катализаторы увеличивают скорость образования продуктов, но не становятся их частью, а остаются отдельными вспомогательными молекулами.
Чтобы произошла химическая реакция между двумя молекулами, эти молекулы должны столкнуться в пространстве. Энергия такого столкновения может быть различной. Если энергии недостаточно для протекания реакции, то молекулы так и останутся в своём первоначальном виде — в виде исходных реагентов, — не прореагируют и не превратятся в продукты реакции. Если же энергия столкновения достаточно высока, то химические связи в исходных молекулах начинают разрываться, вся реагирующая система перегруппировывается, и образуются молекулы продуктов. Минимальное количество энергии, которое необходимо для протекания химической реакции, называется энергией активации. Еще эту величину называют потенциальным барьером реакции. Чем больше энергия активации, тем медленнее протекает химическая реакция, и наоборот — уменьшение потенциального барьера приводит к ускорению химического процесса.
Сам факт наличия энергии активации и ее величина напрямую определяются механизмом химической реакции, то есть последовательностью состояний, через которые протекает превращение реагентов в продукты. На пути от реагентов к продуктам, как правило, существует промежуточное состояние, которое уже не является реагентом (так как химические связи в нем уже начали разрываться), но еще и не является продуктом (так как новые связи еще не сформировались в окончательном виде), и именно поэтому энергия такого состояния больше как энергии продуктов, так и энергии реагентов. Это состояние называется переходным состоянием химической реакции (или активированным комплексом). Энергия активации определяется именно энергией переходного состояния реакции и представляет собой простую разность между энергией переходного состояния и энергией исходных реагентов. Такая разность говорит химику, сколько нужно добавить энергии в реакционную смесь, чтобы реагенты смогли добраться до самой энергетически высокой точки — точки активированного комплекса — и, перевалив через этот барьер, превратиться в продукты (рис. 1).

{kind=link}
Рис. 1. Одна из возможных схем, иллюстрирующих соотношение энергий реагентов, продуктов и переходного состояния для каталитической (красная кривая) и некаталитической (синяя кривая) химических реакций. Изображение © Лаборатория В. Ананикова
Простым и часто используемым наглядным механическим примером переходного состояния и связанной с ним энергии активации является процесс выворачивания наизнанку открытого зонтика под действием налетевшего порыва ветра. Зонтик в нормальном состоянии — это «реагент», и, как бы вы ни крутили в руках этот «реагент», самопроизвольно он не вывернется наизнанку, то есть не превратится в «продукт». Для такого процесса нужна дополнительная энергия — энергия ветра. Под действием энергии воздушного потока зонтик выворачивается наизнанку, и это состояние «продукта» также устойчиво, потому что в нем наш зонтик может находиться сколь угодно долго, если его оставить в покое, и обратно в «реагент» он не превратится. Как можно легко догадаться, «переходным состоянием» этого процесса является «плоский зонтик», который находится на пути от выпуклого зонтика к вогнутому. В этом плоском «высокоэнергетическом» состоянии зонтик находится лишь доли секунды, поэтому оно и остается незамеченным, но именно оно определяет ту энергию, которую нужно затратить на выворачивание. И именно оно служит барьером, отделяющим нормальное состояние зонтика от вывернутого состояния, не давая возможности происходить выворачиванию без дополнительной энергии и позволяя нам пользоваться этой вещью.
Следует отметить, что аналогичные процессы выворачивания происходят не только с зонтиками, но и с молекулами, и называются такие превращения инверсией. Например, молекула аммиака, имеющая пирамидальное («зонтичное»), строение может вывернуться наизнанку, пройдя через плоское переходное состояние, в результате чего образуется неотличимая от первоначальной молекула NH3.
Итак, чтобы реакция началась, молекулы должны столкнуться с достаточной энергией. Для этого частицам необходимо перемещаться с достаточно высокой скоростью. Мерой скорости перемещения молекул служит температура. То есть чем выше температура смеси, тем быстрее перемещаются молекулы в ней, а значит, и сталкиваются они с большей энергией. Отсюда следует простой вывод: для ускорения химической реакции можно просто нагреть смесь. При нагревании доля «быстрых» молекул увеличится и увеличится доля результативных столкновений, то есть актов превращения реагентов в продукт.
Тогда зачем же нужны катализаторы, если ускорять реакцию можно нагреванием? Ответ на этот вопрос заключается в том, что при увеличении температуры начнут протекать побочные процессы, энергия активации которых окажется ниже, чем у целевого процесса, и эти процессы приведут к совсем другим продуктам. Например, при нагревании сложная органическая молекула может разложиться раньше, чем произойдет нужное химическое превращение. Особо важно это для биохимических реакций.
Ферменты — двигатели жизни[]
Известно, что биологические молекулы, в частности белки, способны выполнять свою биологическую функцию только находясь в определенной пространственной форме. Эта форма поддерживается относительно слабыми нековалентными взаимодействиями (например, водородными связями). Даже незначительное увеличение температуры может привести к тому, что потенциальный барьер разрыва этих слабых связей будет преодолен и они диссоциируют, перестав поддерживать форму белковой молекулы. В результате — неправильное функционирование белков и смерть организма. Именно поэтому в природе возникли биологические катализаторы — ферменты, или, как их еще называют, энзимы. Ферменты могут представлять собой белковые молекулы, комплексы белков и нуклеиновых кислот, комплексы белков с металлами. Ферменты катализируют биохимические реакции и позволяют им протекать при низких температурах в живом организме, обеспечивая кроме этого и нужную пространственную форму образующегося продукта, если эта форма важна.
Независимо от того, в какой области встречается катализ — в неорганической химии, органической химии или в биохимии, — суть катализа остается неизменной и сводится к уменьшению энергии активации реакции за счет уменьшения энергии переходного состояния (ведь именно энергии переходного состояния и исходных реагентов определяют потенциальный барьер реакции).
В самых общих чертах этот процесс выглядит так. Катализатор на первой стадии каталитической реакции связывается с исходной молекулой реагента, в результате чего, как правило, происходит активация (упрощенно говоря, ослабление) определенных химических связей. Такие активированные связи легче формируют переходное состояние, а следовательно, и потенциальный барьер реакции уменьшается. Пройдя точку переходного состояния, реагирующая система превращается в продукты, молекулы которых отсоединяются от молекулы катализатора. То есть, завершив каталитический цикл, молекула катализатора снова способна координировать молекулы реагентов, активировать их и ускорять процесс. В этом смысле иногда говорят, что катализатор «не расходуется» в ходе реакции. В нашем примере с зонтиком катализатор должен был бы уменьшать жесткость металлических спиц, чтобы сделать более легким переход в состояние «плоский зонтик».
Начало исследованиям ферментативного катализа было положено еще в XIX веке при изучении процессов брожения. В 1897 году немецкий химик Эдуард Бюхнер (Eduard Buchner) опубликовал свое исследование On alcoholic fermentation without yeast cells (Über alkoholische Gärung ohne Hefezellen), в котором показал, что для сбраживания углеводов не обязательно воздействие живых дрожжевых клеток — достаточно действия некоторых веществ, содержащихся в экстракте, полученном из дрожжевых клеток. Следует отметить, что к этому же выводу за 25 лет до Бюхнера пришла наша соотечественница Мария Михайловна Манасеина. Бюхнер был осведомлен об исследованиях Манасеиной, однако ссылок на ее работы не давал. (См.: V. M. Kovalzon, 1994. Maria Manasseina — a forgotten founder of sleep science и В. М. Ковальзон, 2012. Забытый основатель биохимии и сомнологии.)
До получения этих результатов многие ученые полагали, что живые клетки обладают особой «жизненной силой», и она необходима для работы ферментов. Благодаря исследованиям Бюхнера и Манасеиной было показано, что ферменты — это катализаторы, действие которых аналогично действию хорошо известных катализаторов в неорганической и органической химии, и никакой сверхъестественной «жизненной силы» для ферментативного катализа не требуется. В 1907 году за свое открытие Бюхнер был удостоен Нобелевской премии по химии «за исследования по биохимии и открытие внеклеточной ферментации» («for his biochemical researches and his discovery of cell-free fermentation»).
Позднее, с развитием молекулярной биологии, выяснилось, что функционирование многих ферментов внутри клетки все-таки в той или иной степени отличается от функционирования в выделенном виде. Но связано это с тем, что внутри клетки фермент может связываться с различными клеточными структурами, что меняет его активность, тогда как в чистом виде таких комплексов он, естественно, не образует.
Поскольку молекула фермента, как правило, значительно больше молекулы субстрата (исходного реагента ферментативной реакции), то субстрат связывается только с определенной областью молекулы фермента. Такая область называется активным центром или активным сайтом фермента. Активный центр фермента обеспечивает высокую селективность ферментативной реакции, так как, благодаря уникальному расположению функциональных химических групп в своем составе, он связывается только с молекулой целевого субстрата, тогда как остальные молекулы, присутствующие в клетке, не испытывают сродства к «чужому» ферменту. Собственно, в активном центре фермента и происходит каталитическое превращение субстрата в продукт. Фермент может не только ускорять протекание реакции, но и за счет особенностей строения своего активного центра регулировать пространственную форму образующегося продукта. Эта способность ферментов не менее важна, чем их каталитическая функция, так как биологическая активность многих молекул тесно связана с их пространственной формой.
Как и другие белковые молекулы, молекулы ферментов образованы последовательностью аминокислотных остатков. В водной среде аминокислотная последовательность сворачивается в глобулярную структуру определенным образом, и после этого фермент готов к выполнению своих функций. Данные о структуре многих ферментов могут быть получены с помощью одной из баз структурных данных белков, например Protein Data Bank. В настоящее время известно 5582 фермента (см. Enzyme nomenclature database).
Циклоприсоединение: от цепей к кольцам[]
К началу 2000-х годов накопилось достаточное количество данных, чтобы можно было с уверенностью утверждать: живые организмы способны в своих целях использовать химическую реакцию, считавшуюся ранее исключительно синтетическим методом, а именно реакцию [4+2] циклоприсоединения (рис. 2).

{kind=link}
Рис. 2. Схема реакций [4+2] циклоприсоединения: (а) — межмолекулярная реакция, (б) — внутримолекулярная реакция (буквами R и R’ обозначены в общей форме органические заместители). Изображение © Лаборатория В. Ананикова
Реакция циклоприсоединения (или реакция диенового синтеза), впервые описанная в 1928 году Отто Дильсом и Куртом Альдером (см. O. Diels, K. Alder, 1928. Synthesen in der hydroaromatischen Reihe и M. B. Smith, J. March, 2007. March’s advanced organic chemistry. Reactions, mechanisms, and structure), в настоящее время является одной из наиболее широко используемых реакций в синтетической органической химии (J.-A. Funel, S. Abele, 2013. Industrial Applications of the Diels-Alder Reaction). В 1950 году О. Дильс и К. Альдер были удостоены Нобелевской премии по химии «за открытие и развитие диенового синтеза» («for their discovery and development of the diene synthesis»).
В простейшем случае межмолекулярной реакции Дильса–Альдера (рис. 2а) происходит соединение молекул этилена (диенофил) и 1,3-бутадиена (диен) с образованием циклического продукта — циклогексена. То есть в ходе реакции циклоприсоединения происходит соединение двух непредельных (обладающих ненасыщенными связями) молекул, содержащих 2 и 4 атома углерода, с образованием циклического продукта из шести атомов углерода. Такой тип реакции циклоприсоединения называется [4+2] циклоприсоединением (см. cycloaddition). Реакция Дильса–Альдера может быть и внутримолекулярной, что обеспечивает ее богатые синтетические возможности. В случае внутримолекулярной реакции диен и диенофил принадлежат одной и той же молекуле, а их соединение приводит к продукту сразу с несколькими циклами — полициклическому (рис. 2б).
Диеновым синтезом получают моноциклические и полициклические продукты, которые могут быть как карбоциклическими (то есть содержать в цикле только атомы углерода), так и гетероциклическими (содержать в цикле атомы, отличные от атомов углерода: азот, серу, кислород и другие). Важность реакции циклоприсоединения в значительной степени определяется еще и тем, что в результате не образуются побочные продукты, то есть в этом процессе достигается стопроцентная эффективность «использования» атомов. Данный факт особенно актуален для разработки экологически безопасных химических технологий в контексте развития так называемой «зеленой» химии.
Подавляющее большинство природных соединений содержит в своем составе циклические фрагменты. Реакция циклоприсоединения как нельзя лучше подходит для получения циклических продуктов — как природных, так и искусственных.
Синтетическая реакция циклоприсоединения может быть каталитической (тогда в качестве катализаторов могут использоваться, например, соединения ряда металлов), но иногда для ее проведения не требуется участие катализатора, так как некоторые пары диен/диенофил соединяются и в мягких условиях. Легкость протекания реакции Дильса–Альдера и потребность в катализе для ее проведения зависит от строения диенов и диенофилов (в первую очередь от типа заместителей при двойных связях). Но часто в лабораторных условиях такие химические превращения требуют относительно жестких условий. Например, высоких температур в районе 80–120°C, которые недостижимы внутри живых клеток (если не принимать во внимание немногочисленные экзотические случаи гипертермофильных микроорганизмов, обитающих как раз в этом диапазоне температур). Уникальной особенностью ферментов является их способность обеспечивать протекание реакции с высокой селективностью при намного меньших температурах.
Циклоприсоединение в природе[]
Несмотря на широкое использование диенового синтеза в органической химии, достоверных сведений о протекании реакции Дильса–Альдера в живой природе до конца 1980-х годов не существовало. И вот оказалось, что Природа не оставила без внимания этот удобный способ получения циклических молекул.
В середине 1990-х годов было обнаружено, что препараты на основе культуральных жидкостей некоторых микроорганизмов (которые аналогичны тем, что использовал Бюхнер в своих работах; по-английски их называют cell-free extracts), в частности грибков Alternaria solani, способны катализировать внутримолекулярную реакцию [4+2] циклоприсоединения с образованием соланапиронов (solanapyrones) — фитотоксинов, выделенных в 1983 году (H. Oikawa et al., 1994. First Direct Evidence in Biological Diels-Alder Reaction of Incorporation of Diene-Dienophile Precursors in the Biosynthesis of Solanapyrones и H. Oikawa et al., 1995. Enzymatic activity catalysing exo-selective Diels–Alder reaction in solanapyrone biosynthesis). Ферментативная реакция Дильса–Альдера, катализируемая ферментом соланапиронсинтазой (solanapyrone synthase), обладает высокой стереоселективностью (то есть помимо собственно ускорения реакции обеспечивает и особую пространственную форму продукта), которая труднодостижима традиционными методами синтетической органической химии.
После открытия ферментативного характера реакции образования соланапирона была обнаружена еще одна ферментативная реакция внутримолекулярного циклоприсоединения, в результате которой образуется молекула ловастатина (fungal polyketide lovastatin; см. Поликетиды), найденного в клетках плесневого грибка Aspergillus terreus. В настоящее время в большинстве работ обсуждается механизм образования ловастатина через стадию циклоприсоединения, как наиболее вероятного пути образования этого продукта. Фермент, катализирующий реакцию образования ловастатина, — Lovastatin Nonaketide Synthase, LNKS (см. Поликетидсинтаза) — был выделен в чистом виде, и его каталитическая активность подробно изучена (D. Witter, J. Vederas, 1996. Putative Diels-Alder-Catalyzed Cyclization during the Biosynthesis of Lovastatin; K. Auclair et al., 2000. Lovastatin Nonaketide Synthase Catalyzes an Intramolecular Diels-Alder Reaction of a Substrate Analogue и E. Stocking, R. Williams. Chemistry and Biology of Biosynthetic Diels–Alder Reactions). Оказалось, что роль LNKS во многом заключается в определении стереоспецифичности реакции.
Наиболее изученным ферментом, возможно катализирующим реакцию Дильса–Альдера, на сегодняшний день является выделенная из грибков Macrophoma commelinae макрофомат-синтаза (macrophomate synthase), ускоряющая межмолекулярную реакцию циклоприсоединения с образованием макрофомовой кислоты (macrophomic acid). Этот фермент, содержащий 339 аминокислотных остатков, выделен в виде индивидуального вещества, и его молекулярная структура установлена рентгено-дифракционным анализом (этот физический метод исследования дает возможность определить положения атомов, составляющих молекулу (см.: T. Ose et al., 2003. Insight into a natural Diels–Alder reaction from the structure of macrophomate synthase; G. Pohnert, 2003. Macrophomate Synthase: The First Structure of a Natural Diels-Alderase и T. Ose et al., 2004. Structure of macrophomate synthase).
Следует отметить, что все вышеприведенные ферменты катализируют не только реакцию Дильса–Альдера, но и некоторые превращения, предшествующие стадии циклоприсоединения.
Спинозин: 4 + 2 = 6[]
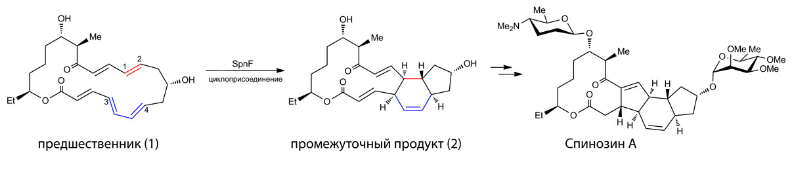
В последнее время был открыт еще один фермент — SpnF, катализирующий реакцию [4+2] циклоприсоединения в процессе образования спинозина А (Spinosyn A) — тетрациклического природного инсектицида (рис. 3), вырабатываемого клетками бактерии Saccharopolyspora spinosa (H. Kim et al., 2011. Enzyme-catalysed [4+2 cycloaddition is a key step in the biosynthesis of spinosyn A]). Этот микроорганизм был обнаружен в почве Виргинских островов в результате исследований, проводимых фармацевтической компанией Элай Лилли (Eli Lilly and Company) в 1980-х годах (F. Mertz, R. Yao, 1990. Saccharopolyspora spinosa sp. nov. Isolated from Soil Collected in a Sugar Mill Rum Still).
Спинозины — это целый ряд соединений, в основе которых лежит тетрациклическая структура, дополненная углеводными заместителями (H. Kirst, 2010. The spinosyn family of insecticides: realizing the potential of natural products research). Такие структуры называются макролидами. По своей биохимической природе спинозин представляет собой вторичный метаболит, а большинство вторичных метаболитов проявляют антибактериальную активность (то есть являются антибиотиками). Однако выяснилось, что спинозин, производимый клетками Saccharopolyspora spinosa, в отличие от других макролидов не обладает выраженной антибактериальной активностью, но проявляет инсектицидную активность, что нехарактерно для макролидных антибиотиков (H. Kirst et al., 1991. A83543A-D, Unique Fermentation-Derived Tetracyclic Macrolides). Поэтому спинозин быстро нашел применение именно в качестве инсектицида.
По сравнению с другими инсектицидами, спинозины быстро разлагаются в окружающей среде, проявляют большую селективность в отношении насекомых-вредителей и меньшую токсичность по отношению к млекопитающим и обитателям водоемов (B. Yano et al., 2002. Spinosad Insecticide: Subchronic and Chronic Toxicity and Lack of Carcinogenicity in Fischer 344 Rats).

{kind=link}
Рис. 3. Структура молекулы спинозина А. Выделена область полициклической системы, образующаяся в результате реакции Дильса–Альдера. Рис. из обсуждаемой статьи в PLoS ONE с адаптацией © Лаборатории В. Ананикова
В клетке бактерии первоначально происходит синтез предшественника спинозина (рис. 4, структура 1), который представляет собой макроциклическую структуру с сопряженными двойными связями (то есть чередующимися с простыми связями).

{kind=link}
Рис. 4. Синтез молекулы спинозина А происходит в результате реакции Дильса–Альдера. Рис. из обсуждаемой статьи в PLOS ONE
Если внимательно посмотреть на структуру предшественника и сравнить со схемой на рис. 2, то станет ясно, что внутримолекулярная реакция [4+2] циклоприсоединения является наиболее оптимальным способом для получения полициклической основы спинозина А. В структуре предшественника можно выделить диеновую часть (окрашена синим цветом) и диенофильную часть (окрашена красным цветом). Реакция циклоприсоединения между этими фрагментами приводит к структуре промежуточного продукта (2) и именно эта стадия катализируется ферментом SpnF. После этого структура (2) в результате ряда превращений (обозначенных удвоенной стрелкой) обретает углеводные заместители и принимает окончательную форму молекулы спинозина. Среди всех этих превращений наибольший интерес исследователей привлекает именно стадия циклоприсоединения, то есть перехода из структуры (1) в (2), потому что в природе реакция [4+2] циклоприсоединения встречается чрезвычайно редко.
SpnF — специалист по циклоприсоединению[]
Интерес именно к этой реакции обусловлен еще и тем, что в отличие от вышеперечисленных ферментов, катализирующих помимо циклоприсоединения и ряд других процессов, SpnF регулирует ход исключительно реакции Дильса–Альдера, то есть Природа «придумала» специальный фермент только для этого процесса.
Механизм действия фермента SpnF до настоящего времени неизвестен. Однако раскрытие этого механизма может иметь фундаментальное значение для расширения наших представлений о ферментативном катализе. Чем же может быть примечательна работа фермента SpnF?
При описании работы ферментов (не обязательно катализирующих процесс циклоприсоединения) принято считать, что их каталитическая роль заключается в разбиении одной стадии с высокой энергией активации на последовательность нескольких стадий с меньшей энергией активации (рис. 5, слева). То есть вместо того, чтобы совершать один большой прыжок через высокий потенциальный барьер из реагентов в продукты, требующий высокой температуры (некаталитическая реакция с переходным состоянием ПС на рис. 5), фермент заставляет реагирующую систему двигаться в сторону продуктов маленькими скачками через низкие барьеры (каталитическая реакция, протекающая через условные переходные состояния ПС1, ПС2, ПС3 на рис. 5 слева). Каждый такой маленький скачок — это отдельное химическое превращение. Между этими маленькими скачками будут образовываться промежуточные продукты (обозначены стрелками, направленными вниз) и только в конце этой последовательности происходит формирование конечного продукта.


{kind=link}
Рис. 5. Общепринятый путь каталитической реакции (слева) и предлагаемый в работе энергетически сбалансированный путь каталитической реакции (справа). ПС — переходное состояние. Изображение © Лаборатория В. Ананикова
Находясь в рамках классической парадигмы ферментативного катализа, мы должны предположить, что для осуществления превращения предшественника (1) в промежуточный продукт (2) фермент SpnF должен разбить эту реакцию на последовательность низкоэнергетических стадий. Почему же это предположение нам не очень подходит? А не подходит оно нам потому, что реакция [4+2] циклоприсоединения в том виде, в каком она изображена на рис. 4, скорее всего, согласованный процесс, то есть он протекает в одну стадию и разбиению на совокупность более мелких стадий поддается с трудом.
Таким образом, в рамках этой общепринятой схемы оказывается невозможным объяснение значительного каталитического эффекта в реакции циклоприсоединения, приводящей к молекуле спинозина А. Чтобы реакция циклоприсоединения из (1) в (2) имела место, фермент должен катализировать только одну эту стадию. Каким же образом он сможет это сделать?
В большинстве работ, обсуждающих гипотетический механизм действия ферментов «природной» реакции Дильса–Альдера, предполагается, что стабилизация переходного состояния стадии циклоприсоединения происходит за счет электростатического взаимодействия вследствие образования водородных связей (H-связей) между субстратом (то есть реагирующей молекулой (1)) и активным центром фермента. Другими словами, за счет образования водородных связей между фрагментами активного центра фермента и молекулой реагента происходит небольшое возмущение распределения электронов в диеновом и диенофильном фрагменте. Поэтому для формирования переходного состояния диену и диенофилу (рис. 1) должно потребоваться меньше энергии, то есть энергия активации реакции должна уменьшиться, чего мы и ожидаем от действия катализатора.
Однако в результате теоретического исследования, выполненного в работе профессора В. П. Ананикова и сотрудников из Института органической химии им. Н. Д. Зелинского РАН, выяснилось, что такое электростатическое влияние со стороны фермента может быть недостаточно для значительного уменьшения энергии активации реакции.
Квантовая химия под давлением[]
Для моделирования химических реакций в деталях, то есть для изучения последовательности всех элементарных стадий процесса и исследования влияния различных факторов на механизм реакции, в последнее время очень часто используются методы квантовой химии. Эти методы позволяют провести расчет энергий всех участвующих в реакции частиц: реагентов, промежуточных и конечных продуктов, переходных состояний, и, следовательно, дают возможность определить все энергетические характеристики реакции, в том числе и ее энергию активации. Квантово-химическое программное обеспечение — это как лаборатория внутри компьютера: можно построить любую новую структуру и определить возможности ее получения или объяснить процессы, приводящие к хорошо известным соединениям. Ограничением здесь являются только вычислительные ресурсы. Современные расчеты методами квантовой химии — это очень ресурсоемкие вычисления, поэтому моделированию с достаточной точностью на широкодоступных рабочих станциях поддаются пока не очень крупные молекулярные системы (несколько сотен атомов). Белковые структуры состоят из сотен, тысяч и десятков тысяч атомов, поэтому для моделирования реакций с участием белков, в частности, ферментов, методами квантовой химии часто приходится прибегать к упрощению модели. (О компьютерном моделировании фолдинга методами классической молекулярной динамики можно почитать здесь.)
Для изучения каталитического действия фермента в реакции образования молекулы спинозина авторы исследования моделировали окружение активного центра несколькими отдельными аминокислотами (одной молекулой глутамина и двумя молекулами серина). В результате формирования водородных связей между молекулами аминокислот и молекулой предшественника (1) происходила некоторая «активация» двойных связей предшественника в отношении процесса циклоприсоединения, и переходное состояние действительно формировалось легче в энергетическом смысле, то есть действие водородного связывания уменьшает барьер реакции. Но это уменьшение оказалось незначительным (1–3 ккал/моль) и не объясняет существенного каталитического эффекта.
Значит, нужно искать другой механизм действия фермента SpnF, обеспечивающий более значительный каталитический эффект фермента.
Тогда исследователи из ИОХ РАН выдвинули гипотезу, в соответствии с которой фермент SpnF захватывает молекулу предшественника (1) в полость или канал, сформированный аминокислотными остатками активного центра. Продвигаясь внутри полости фермента, гибкая молекула циклического субстрата сжимается под действием окружения, в результате чего происходит сближение соединяющихся фрагментов молекулы (диена и диенофила, см. рис. 4). В результате сближения фрагментов структура предшественника (1) приближается к структуре переходного состояния. Поскольку в результате сжатия молекула уже значительно приблизилась к структуре переходного состояния, то для достижения точки переходного состояния потребуется лишь незначительное дополнительное количество энергии (та самая энергия активации). Следовательно, предварительное сжатие субстрата в полости фермента должно значительно уменьшить потенциальный барьер реакции Дильса–Альдера, сохранив ее согласованность, то есть одностадийность.
Но следует понимать, что сжатие молекулы приводит к увеличению ее энергии. Однако это увеличение энергии компенсируется за счет образования водородных связей между молекулой субстрата (1) и аминокислотными остатками в активном центре фермента. Таким образом, двигаясь по каналу фермента, молекула реагента (1) сжимается, и ее энергия растет, но на пути своего следования она встречает аминокислотный остаток, связывается с ним водородными связями и в результате такого связывания энергия всей системы уменьшается, приводя ее к первоначальному значению (напомним, что если у вас были две изолированные молекулы, способные связываться между собой водородными связями, то процесс образования этих связей энергетически выгоден и сопровождается уменьшением полной энергии образующегося комплекса). То есть один акт сжатия/компенсации не приводит к изменению полной энергии системы, потому что эти два процесса меняют энергию в разные стороны. Далее молекула снова сжимается, и ее участки еще ближе придвигаются друг к другу, а увеличение энергии после сжатия снова компенсируется образованием новой водородной связи (без потери старой, образовавшейся на самой первой стадии). И так далее до тех пор, пока структура предшественника (1) не приблизится к структуре переходного состояния. В упрощённой форме этот процесс изображен на рис. 5 справа. «Волны» на кривой каталитического процесса — это и есть циклы сжатия/компенсации: если кривая идет вниз (то есть энергия уменьшается), то это соответствует связыванию с аминокислотным остатком, если энергия увеличивается, то это рост энергии в результате сжатия. После нескольких таких циклов молекула достигает точки переходного состояния (ПС1) согласованной реакции Дильса–Альдера и затем превращается в продукты.
Для количественной характеристики нового гипотетического каталитического механизма рассмотрим рис. 6.


{kind=link}
Рис. 6. Путь реакции для нового каталитического механизма (цветная кривая) характеризуется намного меньшей энергией активации по сравнению с некаталитическим процессом (черная кривая). Рис. из обсуждаемой статьи в PLoS ONE с адаптацией © Лаборатории В. Ананикова
Реакция начинается в точке I, соответствующей изолированным молекулам субстрата (предшественника (1)) и фермента. В результате специфического взаимодействия между ними (то есть образования водородных связей) субстрат располагается в активном сайте фермента (точка II), и в процессе перехода системы из состояния (I) в состояние (II) происходит уменьшение энергии системы на 7,3 ккал/моль за счет образования водородных связей между молекулами фермента и субстрата. Затем происходит сжатие молекулы субстрата до состояния (III), таким образом, что выигрыш энергии, полученный в результате координации первого аминокислотного остатка, компенсируется, и полная энергия системы повышается до уровня энергии системы (I). Далее происходит координация второго аминокислотного остатка (IV), снова сопровождающаяся уменьшением энергии, и последующее сжатие субстрата (V), компенсирующее это уменьшение. В результате третьей стадии координации/сжатия (VI–VII) субстрат геометрически приближается к переходному состоянию (VIII), которое, в свою очередь, связано водородными связями со всеми тремя аминокислотами и его образование уже не требует того количества энергии, как если бы оно формировалось из точки (II).
После того как стадия циклоприсоединения завершилась, продукт остается какое-то время в ассоциированном (связанном) с ферментом состоянии (IX), и лишь затем покидает полость активного сайта (X). В результате энергия активации такого процесса (если отсчитывать ее от самой энергетически низкой точки IV) составляет 19 ккал/моль, что по сравнению с 36,6 ккал/моль для некаталитического процесса (черная линия на рисунке) представляет собой значительное уменьшение энергии активации. Полученный в результате предварительного сжатия молекулы реагента каталитический эффект (около 18 ккал/моль) значительно превосходит каталитический эффект от «обычного» влияния аминокислот (1–3 ккал/моль), рассмотренного выше.
Принципиально важно отметить, что данное превращение проходит всего лишь через одно переходное состояние. В отличие от общеизвестных представлений об энзиматическом катализе, где постулируется обязательное наличие нескольких переходных состояний, предложенная гипотеза позволяет сохранить согласованный механизм стадии циклоприсоединения. Полученный результат развивают наши представления о механизмах работы энзимов и открывают новые возможности для создания каталитических систем с аналогичной последовательностью циклов сжатия/компенсации исходного реагента. Вполне возможно, что предложенный механизм можно распространить и на другие типы реакций, катализируемых ферментами.
В одном из последних номеров журнала Nature Chemical Biology были опубликованы результаты работы, посвященной изучению структуры фермента SpnF (C. Fage et al., 2015. The structure of SpnF, a standalone enzyme that catalyzes [4 + 2 cycloaddition]). Оказалось, что в структуре этого фермента для связывания молекулы субстрата формируется полость объемом 860 кубических ангстрем, который достаточен для включения молекулы субстрата полностью. То есть гипотеза, развиваемая в статье Российских авторов, не лишена некоторых экспериментальных оснований.
Примечания[]
Ссылки[]
Литература[]
- Evgeniy G. Gordeev, Valentine P. Ananikov. Computational Study of a Model System of Enzyme-Mediated [4+2 Cycloaddition Reaction] // PLoS ONE. 2015. DOI: 10.1371/journal.pone.0119984.
| Это незавершённая статья. Вы поможете проекту, исправив и дополнив её.
|
- Страница 0 - краткая статья
- Страница 1 - энциклопедическая статья
- Разное - на страницах: 2 , 3 , 4 , 5
- Прошу вносить вашу информацию в «Ферментативная реакция Дильса–Альдера: на уроке у Природы 1», чтобы сохранить ее